HIGH-THROUGHPUT de novo MITOCHONDRIAL GENOME ASSEMBLY PIPELINE SUPPORTS THE OPTIMAL ENVIRONMENTAL DNA ASSAY DESIGNS IN A GLOBAL CLIMATE CHANGE ERA
Canada is a country with highly rich biodiversity stretching from the Atlantic Ocean to the Pacific Ocean. Biodiversity loss is currently at an unprecedented rate within Canada due to climate change, overfishing, habitat degradation, etc. Development of new environmental monitoring tools for targeted coastal and inland ecosystems is highly necessary. Environmental DNA (eDNA) methods using the genetic material shed by organisms have been applied to monitor at-risk and invasive species across many agriculture and ecology fields. Common eDNA assays include environmental sample collection, followed by DNA extraction and quantitative PCR (qPCR) analysis. This eDNA-based rapid, non-destructive and cost-effective approach helps detect the presence of invasive, at-risk, culturally and economically important species. However, many previously established eDNA qPCR assays lack high-quality primers and this has historically led to poor assay sensitivity and high false positives.
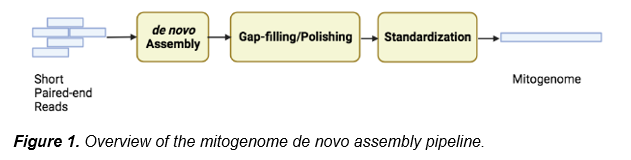
To optimize the eDNA assays, taxa-specific genomic regions need to be characterized. Mitochondrial DNA (mtDNA) is an ideal target for unique genomic sequence identifications as mtDNA have hundreds to thousands of copies in each eukaryotic cell, making it easier to be extracted from the whole-genome shotgun sequencing data through bioinformatics approaches. Due to the high cost for sequencing in the past, the availability of complete mitogenomes in public databases such as National Center for Biotechnology Information (NCBI) is limited and the intra-species variations were not well represented. Hence, we aim to develop a high-throughput de novo mtDNA assembly pipeline that allows the generation of large quantities of standardized high-quality mtDNA genomes which can be later used in capturing unique mtDNA sequences of target taxa. The key steps of the proposed bioinformatics pipeline includes de novo assembly, gap filling/polishing and start-site/strand standardization (Figure 1).