EXPLORING MICROBIAL COMMUNITY DYNAMICS WITHIN RAS USING THREE DIFFERENT SEQUENCING APPROACHES
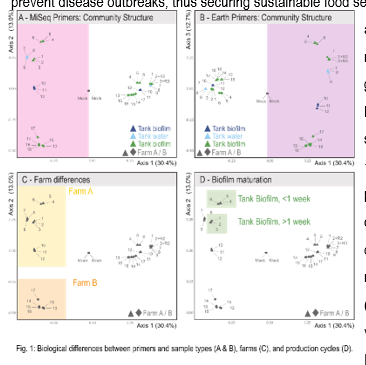
Microbial communities influence farmed animals’ growth, development, and welfare, especially in recirculating aquaculture systems (RAS). Unfortunately, the composition of microbiota communities within most RAS is largely unknown. Establishing accurate, standardized, and user-friendly methods to study microbiota in RAS may lead to RAS systems, better management practices, and natural ways to prevent disease outbreaks, thus securing sustainable food security. We used short- and long-read amplicon sequencing data to compare microbiota classification for two geographically separated, commercial-size RAS fish farms. Since short-read amplicon sequencing targets different regions of the 16S rRNA gene, we conducted a performance comparison between three commonly used primer pairs targeting different hypervariable regions of the 16S rRNA gene (27F/534R: v1-3, 341F/805R (MiSeq): v3-4, and 515F/806R (Earth): v4). In addition, we sequenced the 16S-ITS-23S rRNA regions for comparison (PacBio Shoreline StrainID). The short-read results revealed that samples amplified with the Earth primers resulted in higher read quality, Alpha richness, and Shannon diversity but could not assign as many taxa at the genus level as the MiSeq samples. Even though taxon assignment varied for lower-level taxa, the community composition at the phylum level was similar between primer pairs regardless of sample type (e.g., water vs. biofilm). Both primer pairs could detect biological patterns between samples types (Fig 1A & B), farms (Fig. 1C), and biofilm succession (Fig 1D). The PacBio data could further assign species, allowing for a deeper understanding of functional roles and pathogen detection. We show that 16S rRNA sequencing is sufficient for understanding spatio-temporal patterns in RAS.